Aromatic L-amino acid decarboxylase (AADC) deficiency
What is AADC deficiency?
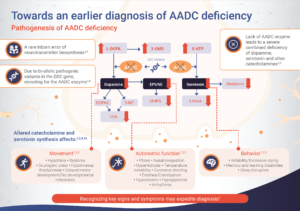
Aromatic L-amino acid decarboxylase (AADC) deficiency is a rare inherited disorder of neurotransmitter synthesis resulting from a mutation in the dopa decarboxylase (DDC) gene.1 Mutations in the DDC gene result in a significant reduction, or complete loss, of AADC enzyme activity, leading to a deficiency in key neurotransmitters, including dopamine, serotonin, epinephrine and norepinephrine.1–3
A reduction in neurotransmitters affects neuronal signaling in the central nervous system, which is required for motor and behavioral development and autonomic function.1,2 AADC deficiency can present with a broad range of symptoms, which usually appear around 3 months of age, and vary from patient to patient.1 The most common symptoms are associated with reduced neurotransmitter levels and include motor and autonomic dysfunction and developmental delay.1,4
What causes AADC deficiency?
AADC deficiency is an autosomal recessive disorder caused by mutations in the DDC gene, resulting in impaired activity of the AADC enzyme.1 The AADC enzyme synthesises several neurotransmitters – such as dopamine, serotonin, adrenaline and noradrenaline – the production of which is affected in patients with AADC deficiency.1
Impaired AADC enzyme activity is also associated with the accumulation of serotonin and dopamine precursors (i.e. 5-hydroxytryptophan [5-HTP], L-3,4-dihydroxyphenylalanine [L-Dopa], and 3-O-methyldopa [3-OMD]) and reduced levels of downstream metabolites (i.e. 5-hydroxyindoleacetic acid [5-HIAA], homovanillic acid [HVA] and 3-methoxy-4-hydroxyphenylglycol [MHPG]).1
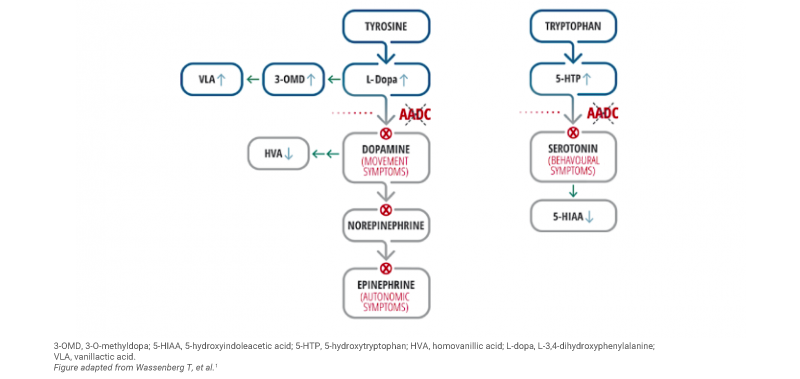
The signs and symptoms of AADC deficiency
AADC deficiency can present with a broad range of symptoms that vary from patient to patient.2,5 Symptoms can appear quite early in life, around 2.7 months of age.1
Key clinical signs and symptoms of AADC deficiency include:1,5
- Hypotonia
- Movement disorders such dystonia and hypokinesia
- Oculogyric crisis
- Developmental delays
- Autonomic dysfunction including ptosis, excessive sweating and nasal congestion
- Diurnal variation – with symptoms worsening toward the end of the day and improving with sleep
The estimated prevalence of AADC deficiency
AADC deficiency is a rare neurometabolic condition. Its prevalence varies widely between different populations, with an estimated global prevalence of 1:26,000 to 1:766,660 live births.6 A founder mutation has been defined in patients of southern Chinese descent, especially from Taiwan and Japan, and the prevalence in Taiwan has been estimated to be 1:66,491 live births (considering only patients born after 2004).1,7
Determining an AADC deficiency diagnosis
AADC deficiency may be challenging to diagnose as patients can present with signs and symptoms that overlap with other, more common conditions, including epilepsy, cerebral palsy or neuromuscular disorders.1,4,10–12
The clinical presentation of AADC deficiency can have some similar signs and symptoms to other neurological conditions (e.g. cerebral palsy, epilepsy) and certain behavioral disorders2
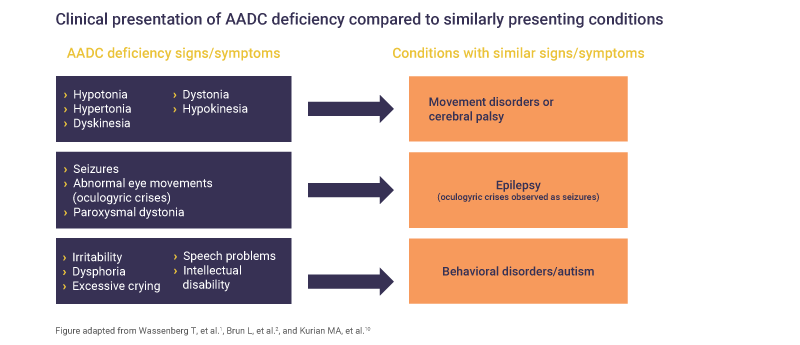
It is recommended that any patient presenting with clinical signs of AADC deficiency should be tested for neurotransmitter disorder.1
Screening for AADC deficiency
Newborn screening provides the opportunity to identify inherited or congenital metabolic or endocrine disorders in newborns.13 There are 2 screening techniques that can be used to identify suspected cases of AADC deficiency:
- Dried blood spot analysis: Elevated 3-OMD concentrations is highly predictive for AADC deficiency and may prompt earlier referral of patients for diagnostic tests14
- Urinary testing: Analysis of urine vanillactic acid to vanillylmandelic acid ratio is also a reliable approach for screening for AADC deficiency.15 If a screening test raises suspicion of an AADC deficiency diagnosis then further confirmation is needed by performing core diagnostic tests.1,8,9
Core diagnostic tests
To confirm a diagnosis of AADC deficiency, a positive genetic test is needed alongside one other positive diagnostic test.1 There are 3 core diagnostic tests that could be performed to confirm an AADC deficiency diagnosis: genetic test, plasma enzyme assay or cerebrospinal fluid (CSF) neurometabolite panel.1
- Gene sequencing: to confirm the presence of a mutation in the DDC gene
- Plasma enzyme assay: a patient with AADC deficiency will have reduced AADC enzyme activity in the plasma
- CSF metabolite panel: a typical CSF panel shows:
- Increased levels of L-Dopa, 5-HTP and 3-OMD
- Reduced levels of HVA, 5-HIAA, and MHPG
- Normal pterins

What is the prognosis of AADC deficiency?
In patients with neurotransmitter disorders such as AADC deficiency, clinical symptoms can be nonspecific and diagnosis is often delayed.23 As a result, patients and carers experience possible differential diagnosis before achieving a definitive diagnosis of AADC deficiency.23
Muscle and joint symptoms can have an impact on many aspects of a patient’s life, including motor function, energy-related symptoms and the ability to communicate.8 Caregivers of patients with AADC report that patients also experience issues with feeding, pain, and impaired cognitive function.8 These symptoms relate to each other and have a wide impact on a patient’s daily activities, emotions, behavior and ability to socialise.8
Patients with AADC deficiency have a substantial burden of living.8
Caregiver burden is high1
Given that most AADC patients have motor and developmental delays, there is a need for life-long care, and one survey found that up to 71% of patients are completely dependent on a caregiver.24 Caregivers report a burden that includes a need to provide 24-hour supervision, administering medication, frequent hospital visits, work disruption and sleep disturbances.24
Caregivers have reported mental tiredness as a result of providing care alongside other mental health symptoms such as depression or anxiety.24
*Retrospective, descriptive, single-center study of patients who received a diagnosis of AADC deficiency at the National Taiwan University Hospital between 2004 and 2016.
Abbreviations
3-OMD, 3-O-methyldopa; 5-HIAA, 5-hydroxyindoleacetic acid; 5-HTP, 5-hydroxytryptophan; AADC, aromatic L-amino acid decarboxylase; CSF, cerebrospinal fluid; DDC, dopa decarboxylase; HVA, homovanillic acid; L-Dopa, L-3,4-dihydroxyphenylalanine; MHPG, 3-methoxy-4-hydroxyphenylglycol.
References
- Wassenberg T, Molero-Luis M, Jeltsch K, et al. Orphanet J Rare Dis 2017;12(1):12.
- Brun L, Ngu LH, Keng WT, et al. Neurology 2010;75(1):64–71.
- Pons R, Ford B, Chiriboga CA, et al. Neurology 2004;62(7):1058–1065.
- Chen PW, Lee NC, Chien YH, et al. Clin Chim Acta 2014;431:19–22.
- Himmelreich N, Montioli R, Bertoldi M, et al. Mol Genet Metab 2019;127(1):12–22.
- Reischl-Hajiabadi A, Okun JG, et al. Mol Genet Metab 2024; 141 (3).
- Hwu WL, Hsu RH, et al. JIMD Rep 2023;64(5):387-392.
- Williams K, Skrobanski H, Werner C, et al. Curr Med Res Opin 2021;37(8):1353–1361.
- Hyland K, Reott M. Pediatr Neurol 2020;106:38–42.
- Krigger KW. Am Fam Physician 2006;73(1):91–100.
- Ng J, Papandreou A, Heales SJ, et al. Nat Rev Neurol 2015;11(10):567–584.
- Kurian MA, Dale RC. Continuum (Minneap Minn). 2016;22(4 Movement Disorders):1159–1185.
- Schulze A, et al. Newborn Screening. In: Sarafoglou K, Hoffman G, Roth K eds. Pediatric Endocrinology and Inborn Errors of Metabolism. 2nd Ed. New York: McGraw-Hill;2017.
- Brennenstuhl H, Kohlmüller D, Gramer G, et al. JIMD 2019;43(3):602–610.
- Brennenstuhl H, Garbade SF, Feyh P, et al. Mol Genet Metab 2020;131(1–2):163–170.
- Evers EA, Sambeth, A, Ramaekers J.G, et al. Curr Pharm Des. 2010;16:1998–2011.
- Chien YH, Chen, PW, Lee NC, et al. Mol Genet Metab. 2016;118:259–63.
- Abdenur JE, Abeling N, Specola N, et al. Mol Genet Metab. 2006;87:48–53.
- Monteleone B and Hyland K. BMC Neurol. 2020;9;20:12. doi:10.1186/s12883-019-1596-8.
- Spitz MA, Nguyen A, Roche S, et al. JIMD Reports 2017;31:85–93.
- Anselm IA and Dallas BT. Pediatr Neurol. 2006;35:142–4.
- Himmelreich N, Bertoldi M, Alfadhel M, et al. Mol Genet Metab. 2023;139:107624.
- Opladen T, Cortès-Saladelafont E, Mastrangelo M, et al. Mol Genet Metab Rep 2016;9:61–66.
- Skrobanski H, Williams K, Werner C, et al. Curr Med Res Opin. 2021;1:1–8.