Duchenne muscular dystrophy (DMD)
What is Duchenne muscular dystrophy?
Duchenne muscular dystrophy (DMD) is a rare, genetic, X-linked recessive neuromuscular disorder that results from mutations in the (DMD) gene that encodes the dystrophin protein.1
DMD, which almost exclusively affects boys, is often characterized by muscle wasting, loss of ambulation, respiratory impairment or cardiomyopathy.1,2 First symptoms typically appear before 3 years of age.3
What is dystrophin?
Dystrophin, encoded by the DMD gene, is responsible for the integrity of muscle structures.1,4 A deficiency of dystrophin leads to progressive muscle degeneration, fibrosis and eventual cardiorespiratory failure.3,4
What causes Duchenne muscular dystrophy?
DMD is caused by mutations in the DMD gene that result in an absent or defective dystrophin protein.1 To date, more than 7000 mutations, including deletions, duplications, and small mutations, have been identified in the DMD gene.5
DMD is an X-linked recessive disorder that primarily affects males1
DMD is a genetic neuromuscular disorder that is inherited in an X-linked recessive manner.1,6 For this reason, boys are almost exclusively affected, however, female carriers are known to have some symptoms based on inactivation of the X chromosome.2,6
Dysfunctional or absent dystrophin leads to muscle degeneration and fibrosis7
Dystrophin is responsible for maintaining the integrity of muscle structures.1,4 Therefore, a DMD mutation can mean that muscle nerve fibers are easily damaged during contractions, which causes inflammation and replacement of fibers by fat and fibrotic tissue; ultimately, leading to loss of muscular function.7

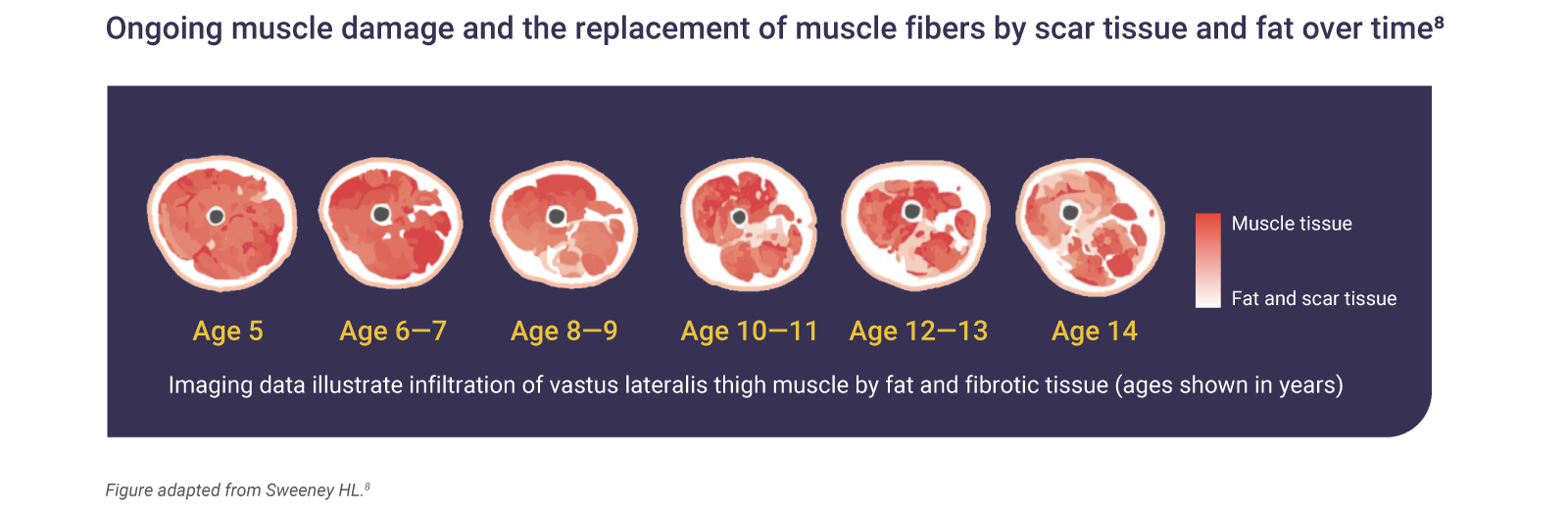
Muscle tissue loss begins in infancy and is irreversible7
The absence of dystrophin in DMD results in ongoing muscle damage and replacement of muscle fibers by scar tissue and fat.7
The incidence and prevalence of Duchenne muscular dystrophy
DMD occurs in 1 in every 3500 to 5000 live male births.2 In ~30% of cases, there is no family history. In female carriers, only ~10% individuals show any disease manifestation.9 Phenotypes for females are usually much milder than in males, although in rare cases, the disease severity can be similar.9
Global DMD prevalence is estimated to be 7.1 per 100,000 males and 2.8 per 100,000 in the global population.10
The signs and symptoms of Duchenne muscular dystrophy
The first symptoms of DMD are normally identified in early childhood by parents. These typically include:1,2
- Delayed motor milestones, e.g. sitting or standing independently
- General motor delays
- Clumsiness and frequent falling
- Gait problems including toe walking
- Flat-footedness
- Delays in walking
- Learning and speech problems
- Cognitive delays
- Changes in serum enzymes
Gowers’ sign, a maneuver used to rise from a supine position in which a child uses their upper arms, pushing on their legs to compensate for weak pelvic muscles, is also a common early sign.11
Identifying motor delays early allows for referral and diagnostic evaluations to take place sooner, allowing for interventions and treatment planning to occur at key developmental milestones.12
Determining a Duchenne muscular dystrophy diagnosis
As DMD is rare, timely diagnosis could be challenging and healthcare professionals may not recognize the signs and symptoms of DMD, resulting in delayed diagnosis.13
The mean age of onset in DMD is 2.5 years, but the average time from reported onset of symptoms to a definitive diagnosis is 2 years with an average age at diagnosis is 4 years.13–15
In patients with no family history of DMD, spontaneous mutations occur in approximately one third of cases; further complicating the diagnostic journey.16
Reasons for diagnostic delay include:17
- Heterogeneity of presentation
- Parental factors: parents may be hesitant in searching for a diagnosis due to conflicting feelings
A first step to diagnosis may be caregiver reports of poor muscle tone, coordination, strength or ambulation as well as slow feeding.13 If developmental concerns are identified then further diagnostic tests are warranted.13
Diagnostic tests
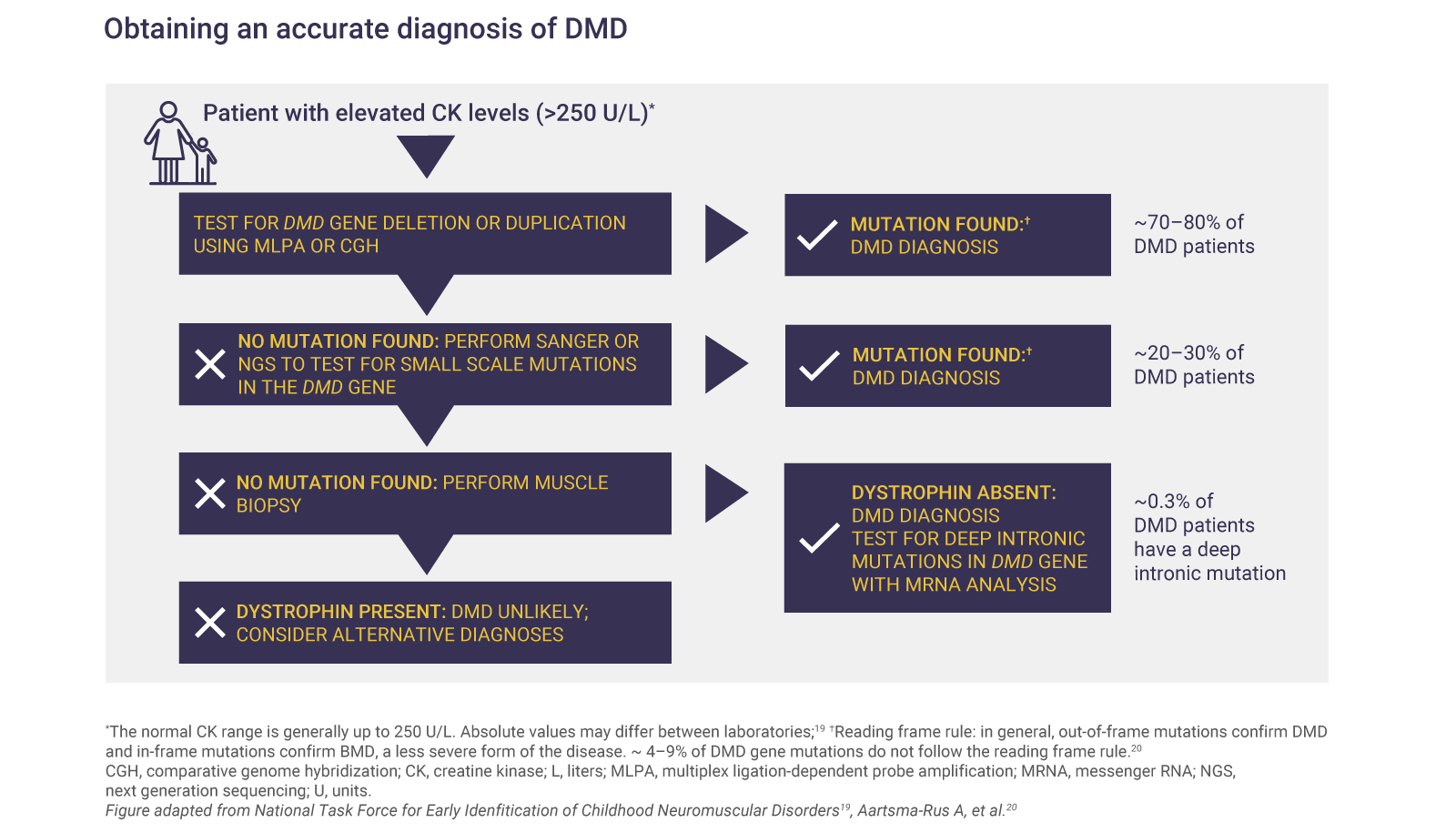
Elevated CK levels reflect muscle damage and are a sign of certain neuromuscular disorders.1,2 A CK test should be ordered if the patient has:1,2
- A positive family history of DMD with suspicion of abnormal muscle function
- Developmental delay, such as difficulty rising to stand or not walking well by 16–18 months
- Unexplained increases in transaminases

Genetic testing can confirm DMD
Genetic diagnosis is required to confirm DMD and to identify the specific mutation causing the disease.1
What is the prognosis for children diagnosed with Duchenne muscular dystrophy?
The natural history of DMD follows a relentless, progressive course during which patients lose functional ability over time.1 Loss of ambulation is a key predictor of disease progression, including severe respiratory insufficiency and death due to respiratory failure.21 Loss of ambulation at a younger age is associated with earlier and more severe respiratory failure when compared to an older age.22

Optimal DMD management involves a coordinated, multidisciplinary team1
Along with improved patient survival, subspecialties caring for DMD patients have shifted to more anticipatory strategies to achieve prevention, earlier diagnosis, and better treatment of predictable and potentially modifiable disease complications.1
Multiple adaptations to daily living are required as a result of DMD.21,24
Environmental accessibility is a key concern as patients begin to experience loss of function in lower limbs, upper limbs, and respiration.21,24
Adaptations to daily living are required beginning at the earliest stages of DMD, which include:1,21
- Changes in diet to help prevent obesity and malnutrition
- Submaximal aerobic activities and stretching to help reduce the risk of contracture and deformity, and minimization of falls to prevent fractures
Transition of care is needed to help patients navigate from pediatric to adult life
Considerations during the transition from pediatric to adult healthcare should include home accessibility and assistive technology, education and vocational planning, and independent transportation to maintain activities of adulthood.21
Patients with DMD are at increased risk of anxiety and depression when compared to those without DMD, especially during points of major care transition.21
Abbreviations
CK, creatine kinase; DMD, Duchenne muscular dystrophy.
References
- Birnkrant DJ, Bushby K, Bann CM, et al. Lancet Neurol 2018;17(3):251–267.
- Matthews E, Brassington R, Kuntzer T, et al. Cochrane Database Syst Rev 2016;(5):CD003725.
- Pichavant C, Aartsma-Rus A, Clemens PR, et al. Mol Ther 2011;19(5):830–840.
- Meyers TA, Townsend D. Int J Mol Sci 2019;20(17):E4098.
- Bladen CL, Salgado D, Foncuberta ME, et al. Hum Mutat 2015;36(4):395–402.
- Kamdar F, Garry DJ. J Am Coll Cardiol 2016;67(21):2533–2546.
- Grounds MD, Terill JR, Al-Mshhdani BA, et al. Dis Model Mech. 2020;13(2):dmm043638.
- Sweeney HL. Developing skeletal muscle MRI/MRS as a biomarker for DMD therapeutic development. Poster presented at the 2014 Annual Connect Conference; 26–28 June, 2014; Chicago, IL.
- Bushby K, Finkel R, Birnkrant DJ, et al. Lancet Neurol 2010;9(1):77–93.
- Crisafulli S, et al. Orphanet Journal of Rare diseases. 2020;15:141
- Darras BT, Urion DK, Ghosh PS. Dystrophinopathies. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. Source Gene Reviews®. Seattle (WA): University of Washington, Seattle; 1993–2019. Published September 5, 2000. Updated April 26, 2018.
- Noritz GH, Murphy NA. Pediatrics 2013;131:e2016–e2027.
- Lurio JG, Peay HL, Mathews KD, et al. Am Fam Physician 2015;91:38–44.
- Ciafaloni E, Fox DJ, Pandya S, et al. J Pediatr 2009;155(3):380–385.
- van Ruiten HJ, Straub V, Bushby K, et al. Arch Dis Child 2014;99:1074–1077.
- Dent KM, Dunn DM, von Niederhausern AC, et al. Am J Med Genet 2005;134:295–298.
- Wong SH, McClaren BJ, Archibald AD, et al. Eur J Hum Genet 2015;23:1294–1300.
- Mendell JR, Lloyd-Puryear M. Muscle Nerve 2013;48:21–26.
- National Task Force for Early Identification of Childhood Neuromuscular Disorders. Child Muscle Weakness. 2019. Available at: http://www.childmuscleweakness.org. Accessed June 2025.
- Aartsma-Rus A, Hedge M, Ben-Omran T, et al. J Pediatr 2019;204:305–313.
- Birnkrant DJ, Bushby K, Bann CM, et al. Lancet Neurol 2018;17(5):445–455.
- Humbertclaude V, Hamroun D, Bezzou K, et al. Eur J Paediatr Neurol 2012;16(2):149–160.
- van Dommelen P, van Dijk O, de Wilde JA, et al. Dev Med Child Neurol 2020;62(10):1198–1204.
- Birnkrant DJ, Bushby K, Bann CM, et al. Lancet Neurol 2018;17(4):347–361.