Hereditary transthyretin amyloidosis
What is hATTR?
Hereditary transthyretin amyloidosis (hATTR) is a severe, progressive, degenerative and fatal disorder that is characterized by the accumulation of amyloid in the body.1–3 With sufficient build-up, amyloid deposits can interfere with normal function, leading to organ failure or death.3
Mutations in the transthyretin (TTR) gene cause deposits of amyloid fibrils that are composed of transthyretin.1–3 Deposit build-up leads to peripheral neuropathy that progress to eventually include ambulation loss.3 Patients can also experience cardiomyopathy, gastrointestinal, ocular or renal dysfunction.3
The disease is progressive leading to death within approximately 10 years (ranging from 5 to 15 years) of symptom onset, depending on many factors.1,4
What causes hATTR?
hATTR is an inherited autosomal dominant genetic disorder, caused by mutations in the TTR gene.5
TTR is a transport protein for thyroxine and vitamin A that is mostly produced in the liver, but some is synthesised in the brain or the retina.1
Mutations in TTR lead to the destabilization and dissociation of TTR tetramers into monomers.5,6 These TTR monomers misfold and form amyloid fibrils that deposit in the peripheral nerves and other organs, leading to peripheral neuropathy and non-specific symptoms.5,6
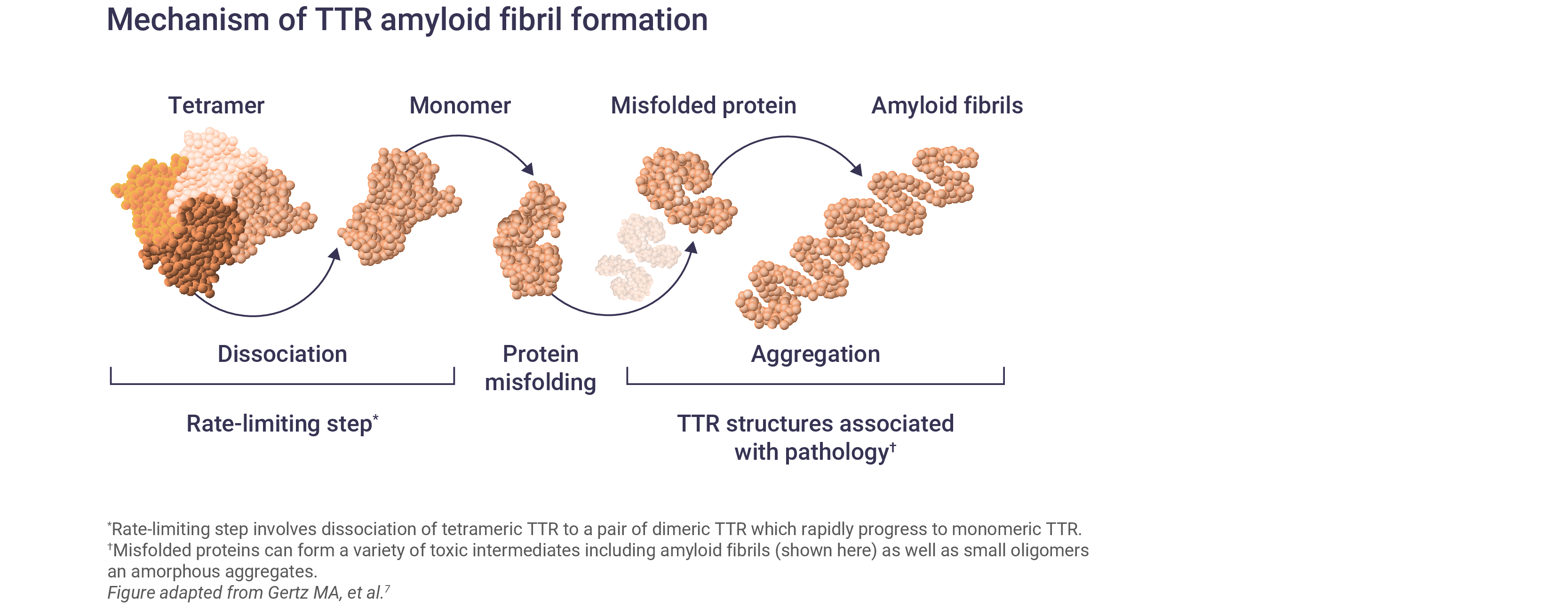
Over 120 TTR variants have been identified, the majority of which are associated with polyneuropathy or cardiomyopathy features; however, most mutations affect multiple organs and there is considerable heterogeneity in disease phenotypes.2,8
The estimated prevalence of hATTR
Worldwide, the estimated prevalence of hATTR is approximately 50,000 people with varying phenotypes.9 Of these, it is thought that 10,000 have hATTR with polyneuropathy while 40,000 have hATTR with cardiomyopathy.10
In the US, hATTR with cardiomyopathy is more prevalent among African Americans, affecting 3–4%.9,10 The polyneuropathy form of hATTR is believed to be endemic to Sweden, Portugal and Japan.9,10
The true prevalence of hATTR could be much higher as the epidemiological data are affected by a lack of awareness, resulting in underdiagnosis compounded by a non-specific clinical manifestation that leads to differential diagnoses of more known, common diseases.11
The signs and symptoms of hATTR
The signs and symptoms of hATTR are numerous and can affect many major organs.12 The majority of TTR mutations are associated predominantly with polyneuropathy or cardiomyopathy; however, most patients with hATTR amyloidosis have mixed clinical phenotypes.2
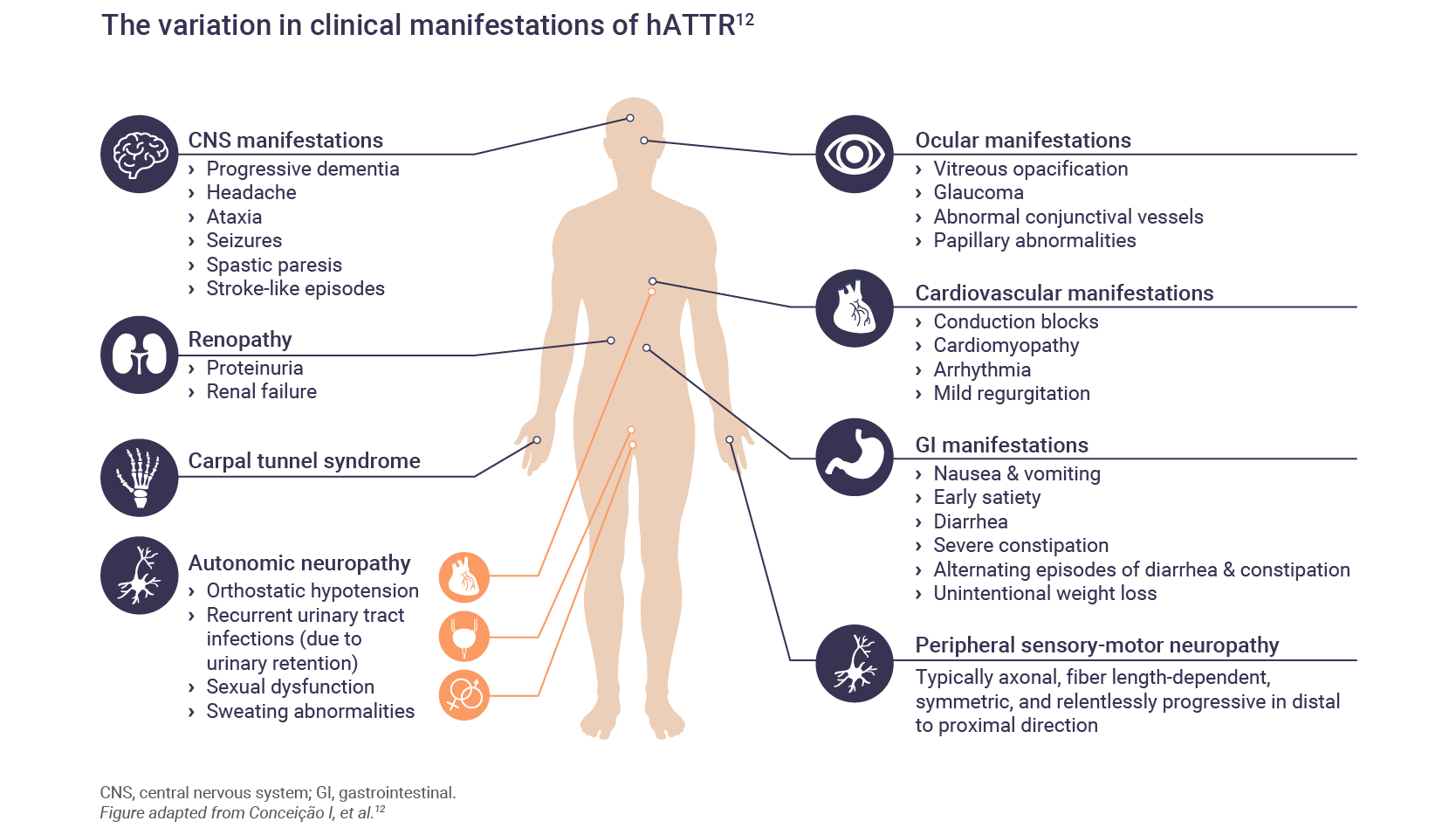
Polyneuropathy
Amyloid deposits can lead to symptoms of peripheral neuropathy where patients may experience sensory difficulties such as paresthesia.3 Symptoms usually begin in the lower limbs and progress proximally.13 Carpel tunnel syndrome is common and may be one of the first symptoms.13
Motor neuropathy can follow with disability of the hands and feet leading to difficulties in walking or dexterity, ultimately leading to a loss of ambulation.3,13
Cardiomyopathy
Cardiomyopathy may be the predominant phenotype or it may develop after the onset of neuropathic symptoms.13 Amyloid deposits can lead to shortness of breath, arrhythmia, diastolic dysfunction, and eventual heart failure.3,13
Gastrointestinal (GI) disturbances
GI symptoms are present in approximately 60% of patients with hATTR.14 In some patients, GI symptoms may be the first noticeable symptom, even before the onset of polyneuropathic symptoms.14 Initial presentation may include constipation, nausea or vomiting.14
In later stages of the disease, diarrhea can become continuous along with incontinence and malnutrition.14
Renopathy
Renal involvement may be rare in hATTR but can include proteinuria, albumineria and/or mild azotemia.1,13
Ophthalmologic manifestations
Since the TTR gene is also expressed in the retina, TTR mutations are also associated with ophthalmologic symptoms.13 Dry eye is an early symptom of hATTR, and as the disease progresses, vitreous opacities and can lead to visual impairment.1,13
CNS manifestations
Symptoms that involve the CNS are rare in hATTR but may include stroke-like symptoms.13
Autonomic neuropathy
Autonomic neuropathy can accompany sensory and motor neuropathy. Autonomic symptoms can include hypotension, constipation or diarrhea, nausea and vomiting, erectile dysfunction, anhydrosis, urinary retention and incontinence.13
Determining a hATTR diagnosis
Patients with hATTR are frequently misdiagnosed, resulting in a significant delay between symptom onset and diagnosis.15 Lack of disease awareness and overlap of symptoms with other common conditions, such as hypertension and diabetes, hampers the diagnosis of hATTR.16
The resulting delay in diagnosis and misdiagnosis can negatively impact treatment approaches and outcomes.16 Therefore, establishing early diagnosis requires increased awareness.16
Red-flag symptom clusters may warn of hATTR diagnosis.5,12 Patients presenting with progressive, symmetric sensory length-dependent polyneuropathy or autonomic dysfunction, with one or more of the red flag signs and symptoms, should be tested for hATTR5,12,17:
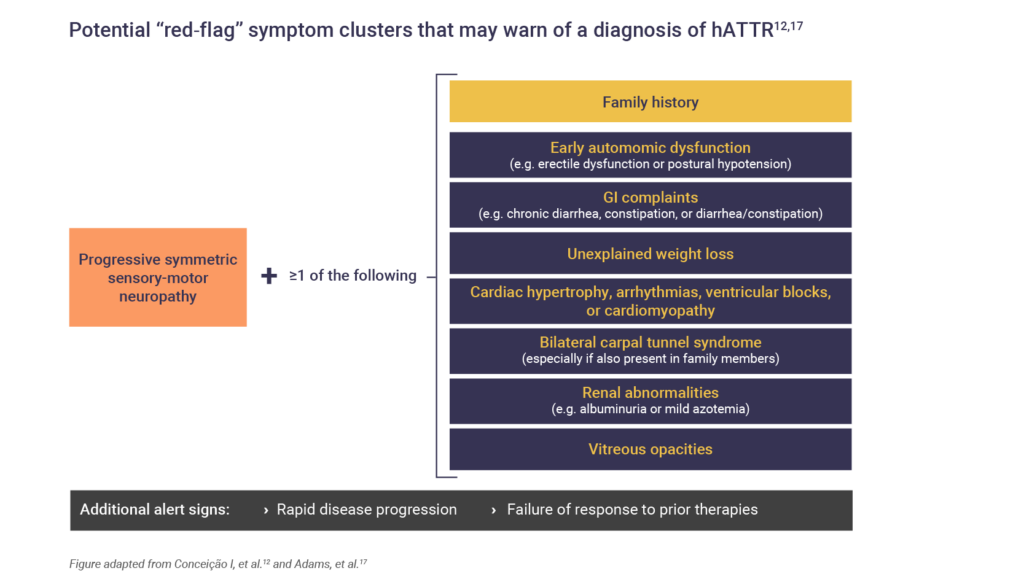
Neurologic and physical examinations for hATTR
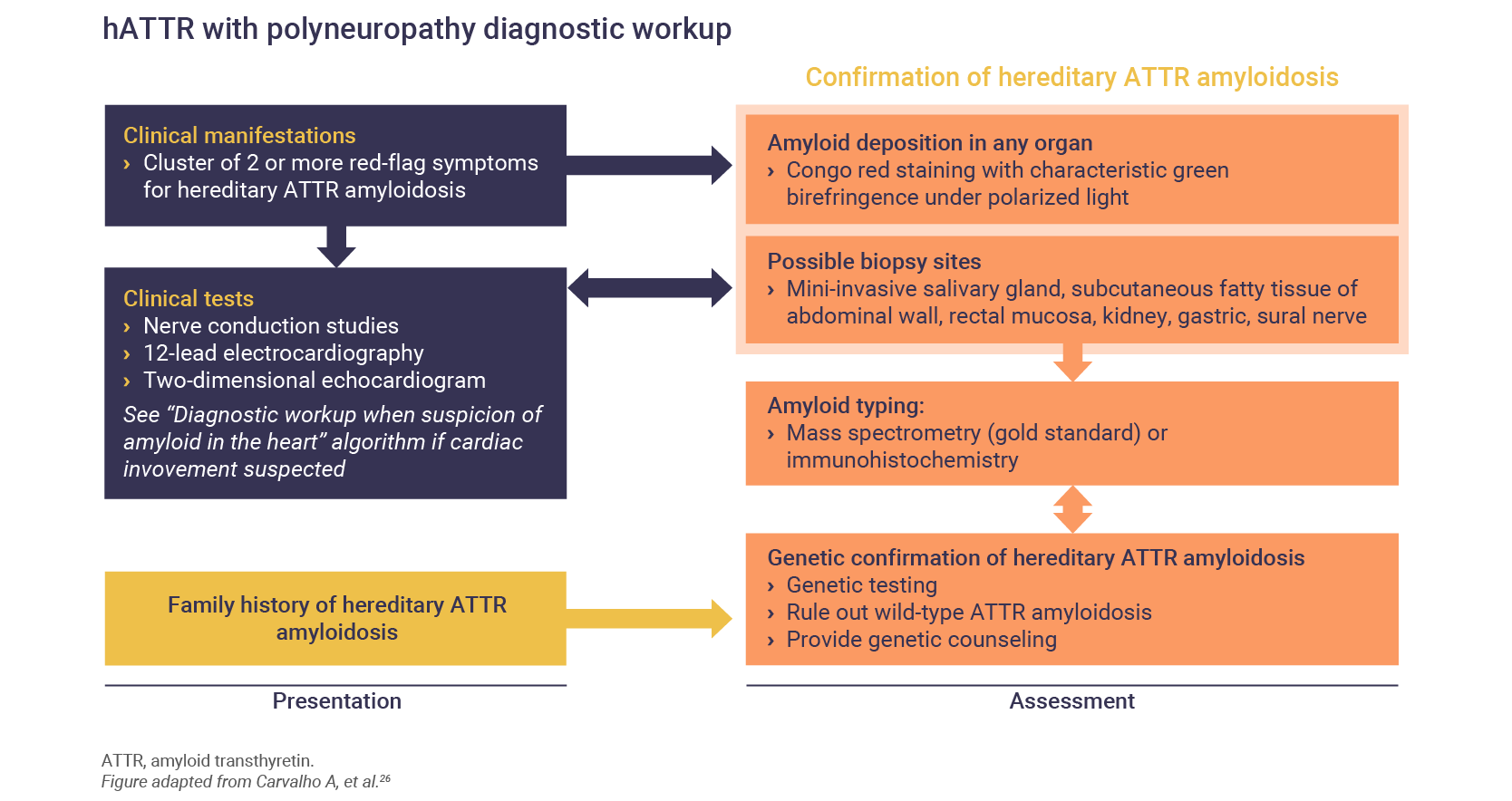
The diagnosis of hATTR involves neurologic and physical examinations, and several different types of diagnostic tests, including:
- Genetic testing and DNA sequence analysis of the TTR gene5,17
- Cardiac tests17:
- Echocardiography18
- Electrocardiogram (ECG)17,18
- Scintigraphy17–19
- Laboratory tests for cardiac biomarkers (troponin B-type natriuretic peptide)20,21
- Cardiac magnetic resonance (CMR) imaging18
- Neurophysiological tests:
- Electrodiagnostic (EDx) testing including nerve conduction studies (NCS), needle electromyogram (EMG), and a variety of special studies22
- Quantitative Sensory Testing (QST)23
- Quantitative Sudomotor Axon Reflex Testing (QSART)24
- Sympathetic Skin Response (SSR)25
- Biopsy1,17
Following cardiac and neurologic testing, patients presenting with signs and symptoms of hATTR should undergo a tissue biopsy to confirm the presence of amyloid deposits.1
Algorithms for the diagnosis of both the polyneuropathy of cardiomyopathy of hATTR have been published26:
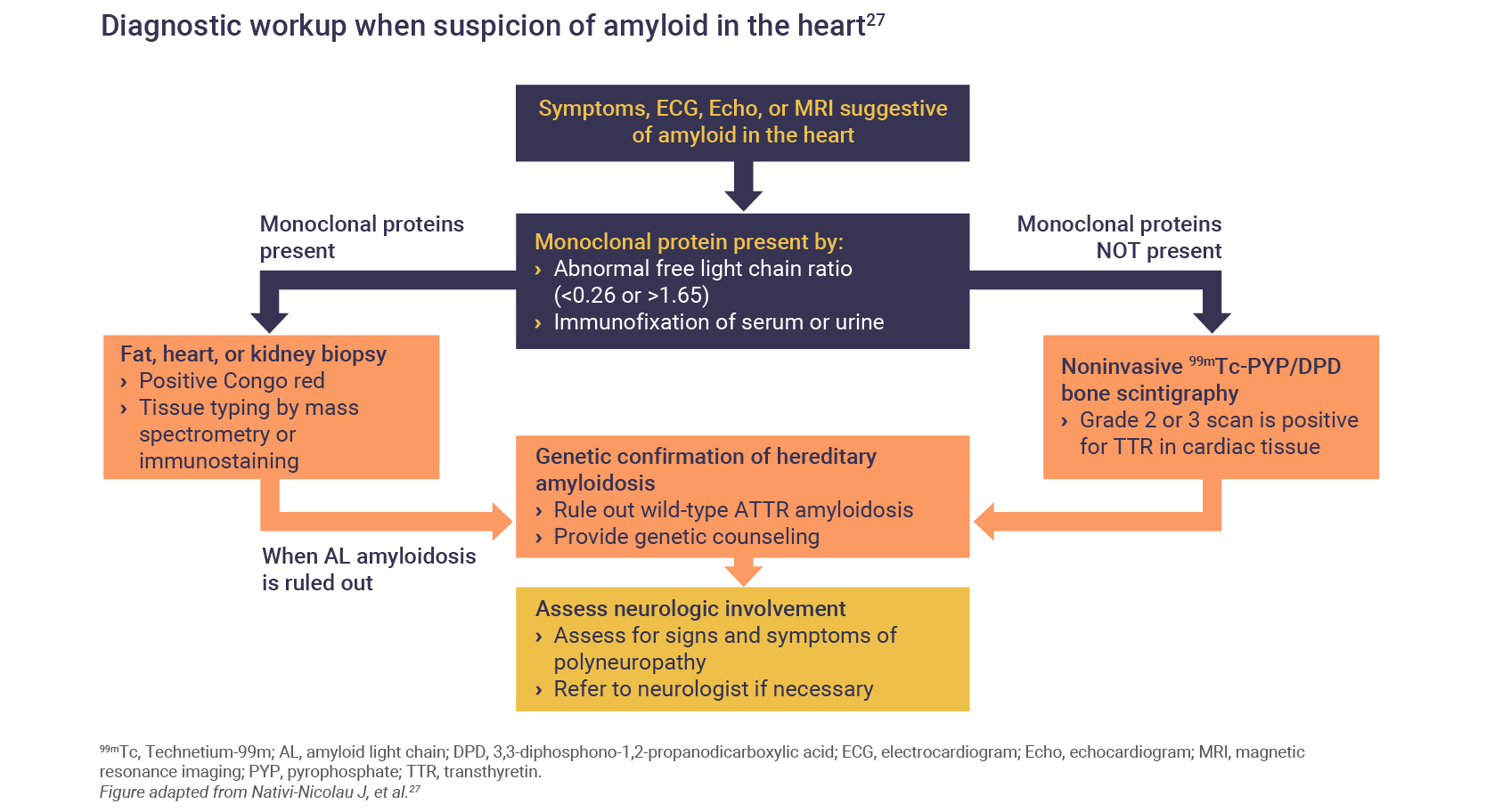
What is the burden of hATTR in patients?
Disease burden is high for patients with hATTR. The clinical manifestations impact multiple aspects of daily life, and the burden of disease increases rapidly as the disease progresses.3
An analysis of neuropathy‐related QoL found that patients with hATTR amyloidosis experience:
- A disease burden equivalent to that of patients with type 2 diabetes who have diabetic neuropathy accompanied by a history of ulceration, gangrene, or amputation3
- The burden on physical functioning for patients with hATTR amyloidosis was worse than that of patients with Crohn’s disease, irritable bowel syndrome, and diabetic neuropathy, while comparable to that of patients with congestive heart failure and multiple sclerosis3
Patients with hATTR amyloidosis often struggle to work and complete household chores due to their symptoms. Many patients report being unable to work, or report challenges attending work on a consistent basis.28
Findings from questionnaires completed by caregivers of patients with hATTR report substantial burden on caregivers, including work impairment, substantial time spent on providing care (mean 45.9 h/week), and poor mental health.29
Given the progressive and multisystemic nature of hATTR amyloidosis, the wide variety of clinical presentations often leads to misdiagnosis and diagnostic delays.30 This incorrect or delayed diagnosis severely impacts patient quality of life as a consequence of the progressive nature of hATTR amyloidosis that leads to significant disability and mortality.28,30
Abbreviations
CMR, cardiac magnetic resonance; CNS, central nervous system; ECG, electrocardiogram; EDx, electrodiagnostic; EMG, electromyogram; GI, gastrointestinal; hATTR, hereditary transthyretin amyloidosis; NCS, nerve conduction studies; QoL, quality of life; QSART, Quantitative Sudomotor Axon Reflex Testing; QTS, Quantitative Sensory Testing; SSR, Sympathetic Skin Response.
References
- Ando Y, Coelho T, Berk JL, et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis 2013;8(1):1–18.
- Luigetti M, Romano A, Di Paolantonio A, et al. Ther Clin Risk Manag 2020;16:109–123.
- Yarlas A, Gertz MA, Dasgupta NR, et al. Muscle & Nerve 2019;60(2):169–175.
- Baker KR, Rice L. Methodist Debakey Cardiovasc J 2012;8(3):3–7.
- Sekijima Y, Ueda M, Koike H, et al. Orphanet J Rare Dis 2018;13(1):1–17.
- Buxbaum JN, Reixach N. Cell Mol Life Sci 2009;66(19):3095–3101.
- Gertz MA, Mauerman ML, Grogan M, Coelho T. Brain and Behaviour 2019;9:e01371
- Maurer MS, Hanna M, Grogan M, et al. J Am Coll Cardiol 2016;68(2):161–172.
- Picken MM. Acta Haematol 2020;143(4):322–334.
- Hawkins PN, Ando Y, Dispenzeri A, et al. Ann Med 2015;47(8):625–638.
- Schmidt HH, Waddington-Cruz M, Botteman MF, et al. Muscle & Nerve 2018;57(5):829–837.
- Conceição I, González-Duarte A, Obici L, et al. J Peripher Nerv Syst 2016;21(1):5–9.
- Coelho T, Ericzon B, Falk R, et al. A guide to transthyretin amyloidosis. 2018. Available at https://amyloidosis.org/sites/default/files/pdf-docs/pages/resources/2023-03/2018 ATTR.pdf Accessed June 2025.
- Wixner J, Mundayat R, Karayal ON, et al. Orphanet J Rare Dis 2014;9(1):1–9.
- Cortese A, Vegezzi E, Lozza A, et al. J Neurol Neurosurg Psychiatry 2017;88(5):457–458.
- Vaxman I, Gertz M. Acta Haematol 2020;143(4):304–311.
- Adams D, Ando Y, Beirão JM, et al. J Neurol 2021;268:109–2122
- Siddiqi OK, Ruberg FL. Trends Cardiovasc Med 2019;28(1):1–28.
- Cytawa W, Teodorczyk J, Lass P. Pol J Radiol 2019;79:222–227.
- Gertz MA, Benson MD, Dyck PJ, et al. J Am Coll Cardiol 2015;66(21):2451–2466.
- Mythili S, Malathi N. Biomed Rep 2015;3(6):743–748.
- Sonoo M, Menkes DL, Bland JDP, et al. Clin Neurophysiol Pr 2018;3:78–88.
- Mücke M, Cuhls H, Radbruch L, et al. Schmerz 2021;35(Suppl 3):153–160.
- Novak P. Quantitative autonomic testing. J Vis Exp 2011;53:1–24.
- Sympathetic Skin Response. Vitalscan. Available at: https://www.vitalscan.com/dtr_sudo_ssr.html Accessed June 2025.
- Carvalho A, Rocha A, Lobato L. Liver Transpl 2015;21(3):282–292.
- Nativi-Nicolau J, Maurer MS. Curr Opin Cardiol 2018;33(5):571–579.
- Lovley A, Raymond K, Guthrie SD, et al. J Patient Rep Outcomes 2021;5(3):1–10.
- Stewart M, Shaffer S, Murphy B, et al. Neurol Ther 2018;7(2):349–364.
- Gertz MA. Am J Manag Care 2017;23(7 Suppl):S107–S112.