Familial chylomicronemia syndrome (FCS)
What is familial chylomicronemia syndrome?
Familial chylomicronemia syndrome (FCS) is a rare, autosomal recessive disorder of chylomicron metabolism that results from loss-of-function mutations within the genes that encode key checkpoint molecules in lipolysis.1 FCS is characterized by impaired clearance of triglyceride (TG)-rich lipoproteins from plasma, leading to severe hypertriglyceridemia (HTG [TG levels >10 mmol/L] [>885 mg/dL]) and the abnormal persistence of chylomicrons in fasting plasma.1–3
What are chylomicrons?
Chylomicrons are large, lipoprotein particles produced after a meal. Lipoprotein lipase (LPL) clears chylomicrons from the plasma by hydrolysing the triglycerides into fatty acids and glycerol.1 In FCS, chylomicron clearance from plasma is impaired due to genetic mutations that affect LPL enzyme activity.1
FCS onset is typically during childhood or early adulthood, usually presenting with episodes of abdominal pain and recurrent acute pancreatitis (AP).1 It is often associated with failure to thrive.3,4 Approximately 25% of affected children develop symptoms before the age of 1 and the majority develop symptoms before 10 years of age.4
FCS nomenclature
Chylomicronemia is the accumulation in the bloodstream of chylomicrons.1 “Chylomicronemia syndrome” refers to the presence of at least 1 clinical feature accompanying primary chylomicronemia, such as eruptive xanthomas, lipemia retinalis, pancreatitis, or hepatosplenomegaly.2
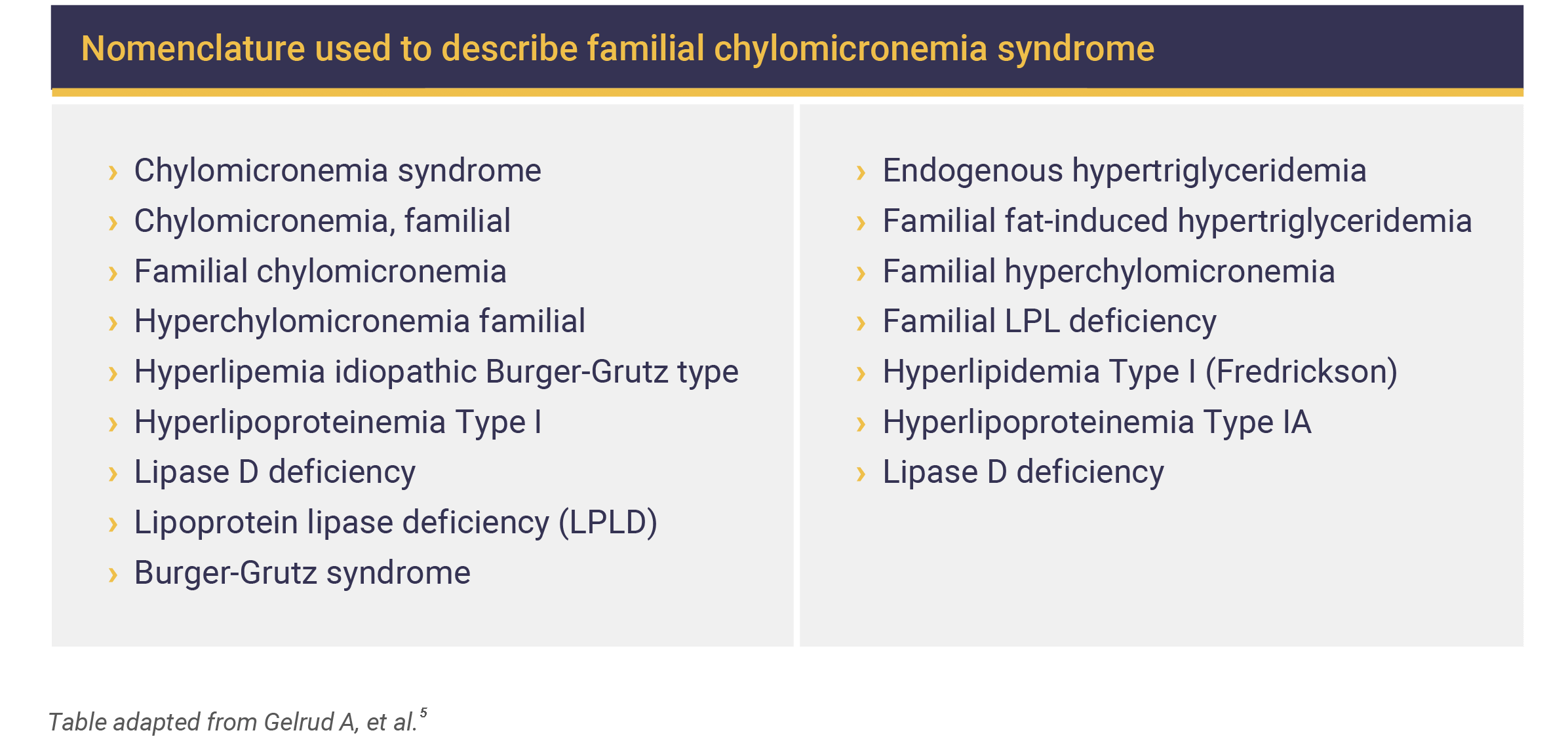
FCS may have different names, such as:5
What causes FCS?
FCS is an autosomal recessive genetic condition resulting from mutations in the LPL gene. These mutations affect the activity of the LPL enzyme, resulting in accumulation of chylomicrons in the plasma.3,6 Mutations in the LPL gene account for more than 80% of FCS cases, and more than 180 mutations have been identified.6
Some cases of FCS may be caused by mutations in 4 additional genes that are involved in LPL function, namely APOC2, APOA5, LMF1, and GPIHBP1. These genes encode apolipoprotein (apo) C-II and A-V, lipase maturation factor 1 (LMF1), and glycosylphosphatidylinositol-anchored high-density lipoprotein (HDL)-binding protein 1 (GPIHBP1).2,7,8 These genes and their associated cofactors modify the hydrolysis of chylomicrons. Recessive mutations in these genes may cause symptoms that are less severe and have a late onset.2
Immunogenic causes of FCS
There has been 1 instance of a patient with autoantibodies against LPL reported in 1989, but the frequency of immunologic FCS is rare, with few cases reported since.3
The estimated prevalence of FCS
FCS is a rare disease, with an estimated worldwide prevalence of approximately 1 individual per million. It has been described in all ethnicities, although a higher prevalence has been observed in some geographical areas such as Quebec, due to a founder effect.1,2
The signs and symptoms of FCS
Physical symptoms of FCS
Patients with FCS can present with varied signs and symptoms including abdominal pain, a failure to thrive, nausea and vomiting, eruptive xanthomas, lipemia retinalis, and hepatosplenomegaly.4
Neurological and cognitive symptoms such as irritability, memory problems, and depression are often documented in patients with FCS. Patients may also present with low body weight, often caused by poor food intake due to abdominal pain.2
AP is a severe and life-threatening complication of FCS and recurrent AP may occur in up to 50% of patients with FCS.8 AP may lead to chronic pancreatitis, permanent damage to pancreatic tissue, pancreatic insufficiency, and type 2 diabetes.4
Clinical symptoms of FCS
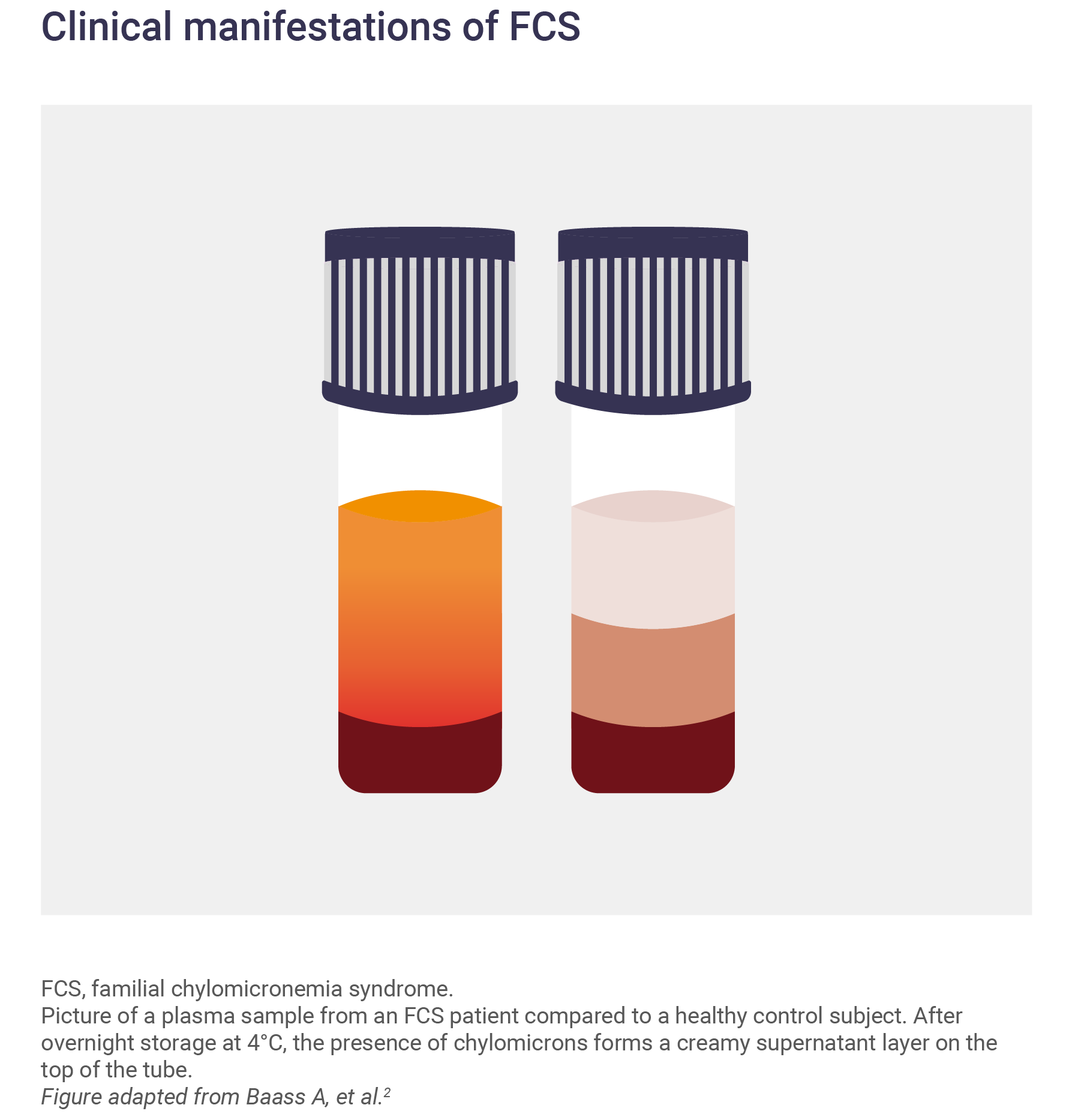
FCS is characterized by persistent chylomicronemia with high plasma TG levels (>1,000 mg/dL), even after a period of fasting.3,7
In a fasting plasma sample, the presence of chylomicrons is indicated by a ‘creamy’ appearance. After resting, the chylomicrons form a white layer on the top of the tube.3
Determining a diagnosis of FCS
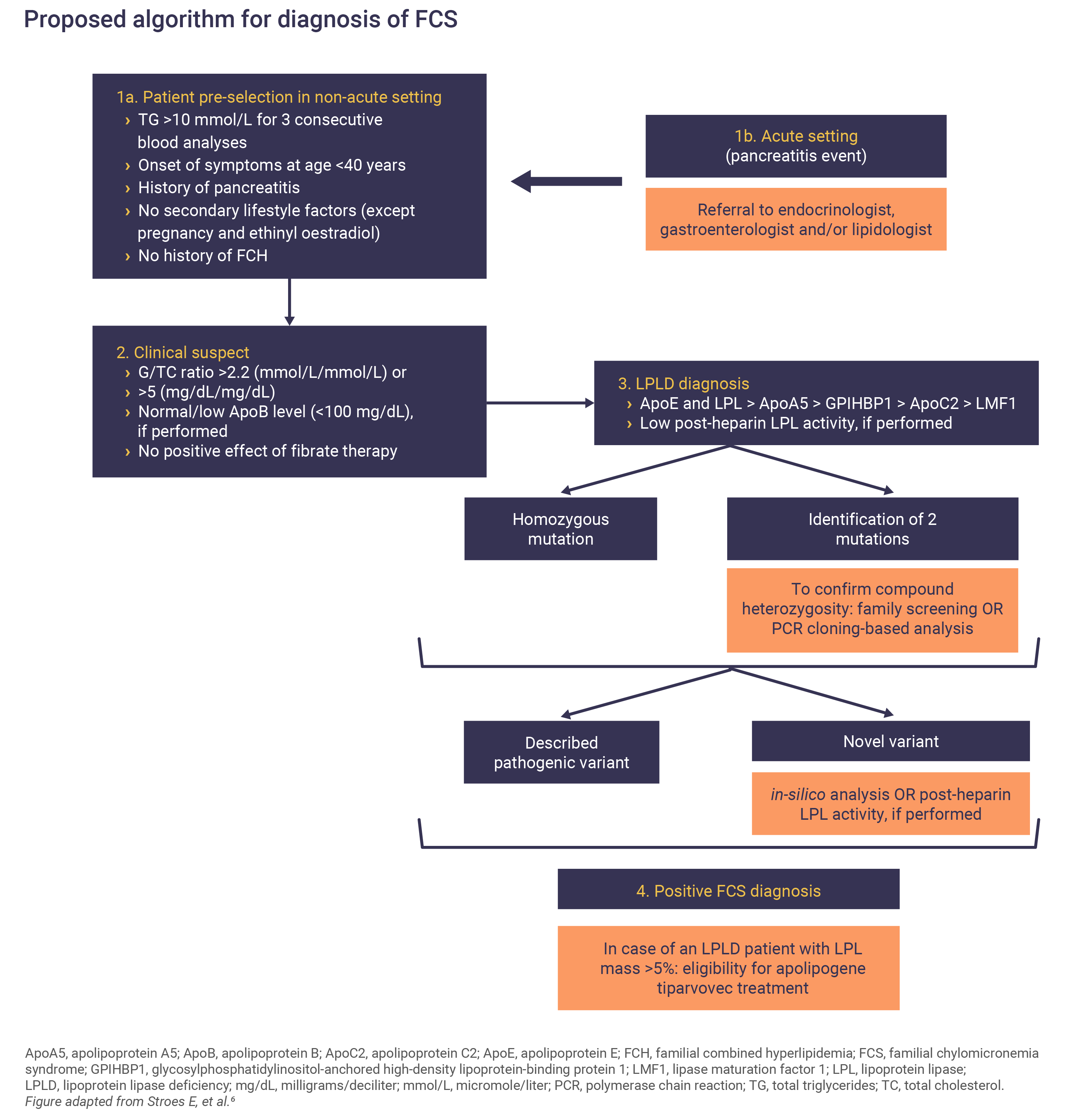
FCS symptoms usually appear early on in life; however, because of its rarity FCS diagnosis is often delayed until later in childhood, or even adulthood.3
A multidisciplinary team approach, including general practitioners, endocrinologists, gastroenterologists and lipid experts and increased awareness of FCS is key to timely diagnosis.6
Proposed scoring system for FCS
Because FCS is a rare condition, it could be underdiagnosed. In this model, each clinical variable is given particular weighting. Variables include fasting TG values, abdominal pain, age of symptom onset, response to lipid-lowering therapies and history of pancreatitis or hyperlipidemia. The model scores indicate the likelihood of a patient having FCS.2,3

FCS should be suspected in patients with fasting severe HTG, i.e. TG ≥885mg/dL, in the absence of secondary causes such as obesity, alcohol, diabetes, or other diseases. FCS patients don’t respond to lipid-lowering agents and an absence of response should raise suspicion of FCS.6 Fasting TG levels remain extremely elevated at different time points and finding raised TG levels in several samples should prompt testing for a genetic disease.6
Although AP is the most serious clinical manifestation of FCS, not all patients suffer from pancreatitis events and this should not exclude its suspicion if other signs or symptoms are indicative.6
Confirming an FCS diagnosis
Genetic sequencing of LPL, APOC2, GPIHBPI, APOA5 or LMF1 to confirm the presence of a mutation is required to confirm an FCS diagnosis.3,6
What is the burden of FCS?
Patients with FCS experience significant clinical and psychosocial burdens that reduce their quality of life and limit employment and social interactions. In one study, only 22% of FCS patients reported being fully employed, and 75% attributed FCS as a major factor responsible for their unemployment status.9
Strict dietary adherence is critical for FCS patients of all ages, but is difficult to maintain and is a source of a significant burden.10 Managing the lifestyle events involved with a restricted fat intake is draining and time-consuming for almost all patients and can be associated with fear, anxiety, helplessness, and guilt when dietary fat limits are exceeded.8
Almost two-thirds of patients with FCS reported that their condition significantly affected their mental and emotional well-being by impacting their self-worth.8 Many patients also expressed concerns regarding the prognosis of their condition and how this may affect their future health and employability.8
Abbreviations
AP, acute pancreatitis; apo, apolipoprotein; ApoA5, apolipoprotein A5; ApoB, apolipoprotein B; ApoC2, apolipoprotein C2; ApoE, apolipoprotein E; FCH, familial combined hyperlipidemia; FCS, familial chylomicronemia syndrome; GPIHBP1, glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1; HDL, high-density lipoprotein; HTG, hypertriglyceridemia; LMF1, lipase maturation factor 1; LPL, lipoprotein lipase; LPLD, lipoprotein lipase deficiency; PCR, polymerase chain reaction; TC, total cholesterol; TG, triglyceride.
References
- Brahm AJ, Hegele RA. Nat Rev Endocrinol 2015;11:352–362.
- Moulin P, Dufour R, Averna M, et al. Atherosclerosis 2018;275:265–272.
- Baass A, Paquette M, Bernard S, et al. J Intern Med 2020;287:340–348.
- Burnett JR, Hooper AJ, Hegele RA. Familial Lipoprotein Lipase Deficiency. In MP Adam, HH Ardinger, RA Pagon, et al., editors. GeneReviews® – NCBI Bookshelf. Seattle, WA: University of Washington, Seattle; 1993–2021. http://www.ncbi.nlm.nih.gov/books/NBK1308/
- Gelrud A, Williams KR, Hsieh A, et al. Expert Rev Cardiovasc Ther 2017;15:879–887.
- Stroes E, Moulin P, Parhofer KG, et al. Atheroscler Suppl 2017;23:1–7.
- Hegele RA, Berberich AJ, Ban MR, et al. J Clin Lipidol 2018;12:920–927.
- Davidson M, Stevenson M, Hsieh A, et al. J Clin Lipidol 2018;12:898–907.
- Falko JM. Endocr Pract 2018;24:756–763.
- Williams L, Rhodes KS, Karmally W, et al. J Clin Lipidol 2018;12:908–919.