What is Huntington’s disease?
Huntington’s disease is a rare, hereditary, degenerative disorder of the brain characterized by a combination of motor, behavioral, and cognitive disturbances.1,2 Although Huntington’s disease onset is variable, symptoms usually begin between the ages of 35 and 55 years.3 It can also begin in childhood or early adulthood, and is then known as Juvenile Huntington’s disease.3,4
What causes Huntington’s disease?
Huntington’s disease is an autosomal dominant inherited disorder caused by a mutation in the HTT gene that encodes the huntingtin protein.2 The single mutation in HTT is an expansion of trinucleotide cytosine, adenine, and guanine (CAG) repeats.2 Unaffected individuals have around 20 CAG repeats in HTT, but those with Huntington’s disease can have 40 or more.5
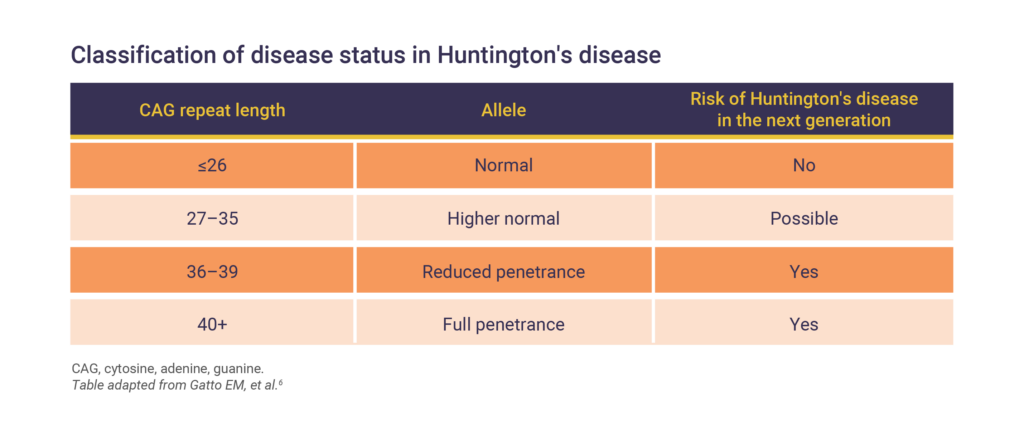
This alteration in the HTT gene results in a variety of problems affecting:6
- Brain development
- Regulation of the transcriptional process
- Balance of histone acetylation/deacetylation and glial activation
- Mitochondrial function
- Axonal transport of organelles by microtubules
- Regulation of signaling pathways
- Multimerisation of mutant HTT
- Autophagy
The estimated incidence and prevalence of Huntington’s disease
Following the introduction and wide implementation of genetic testing for Huntington’s disease, the accuracy of prevalence estimates has improved and resulted in several populations with substantially higher prevalence than previously thought.7
Global incidence has been estimated as 0.48 cases per 100,000 person‐years with geographical variance and significantly higher incidence of Huntington’s disease in Europe and North America compared with Asia.
The global prevalence of Huntington’s disease is estimated to be 4.88 per 100,000.8 Geographical variance has been demonstrated, with prevalence being significantly higher in North America and Europe compared with Africa.8
The signs and symptoms of Huntington’s disease
Signs and symptoms of Huntington’s disease vary widely from person to person, and between stages of the disease.9,10
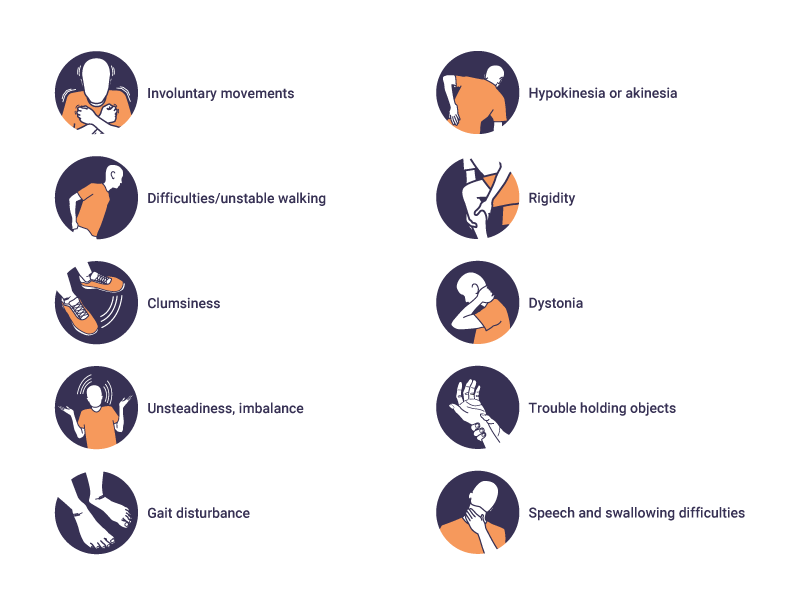
Motor symptoms
The onset of motor symptoms can occur at any age, but on average emerges between ages 35 and 44 years.2,9 Motor symptoms are a mixture of chorea, bradykinesia, and dystonia, which affect posture, balance, and gait.2,3 These symptoms start in the distal extremities such as fingers and toes, before spreading to all other muscles in the body. Motor symptoms include:10
Cognitive symptoms
Cognitive symptoms of Huntington’s disease may be evident more than 15 years before motor diagnosis and develop gradually, eventually being widespread across faculties at the later stages of the disease. These include:2,11–14
- Memory loss
- Dementia
- Poor attention
- Slow psychomotor functions
- Slow processing speed
- Poor executive functions
Neuropsychiatric symptoms
Psychiatric symptoms often appear before motor symptoms and are typically present from the early stages of the disease.10 These can include:2,10–12
- Agitation
- Irritability
- Disinhibition
- Depression
- Apathy
- Anxiety
- Delusions
- Hallucinations
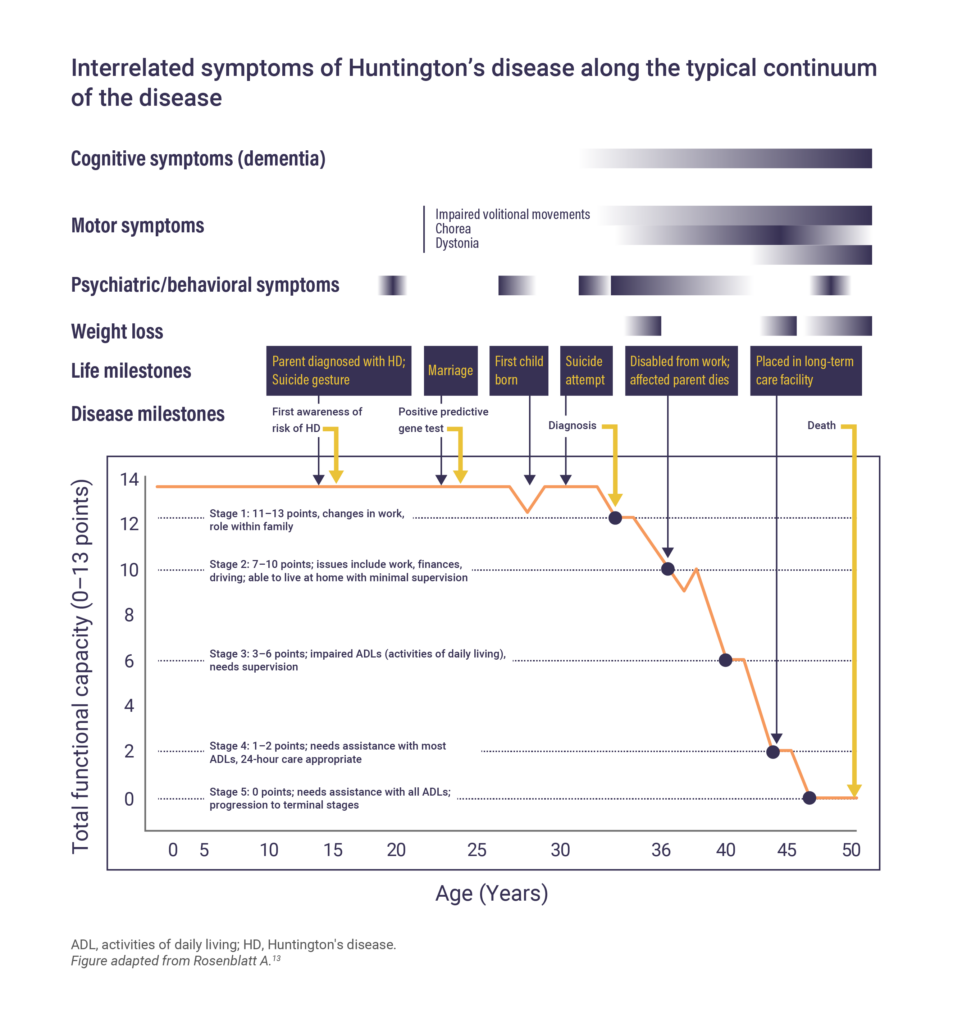
Other less prevalent and often debilitating symptoms of Huntington’s disease include weight loss, sleep and circadian rhythm disturbances, and autonomic nervous system dysfunction.10 It is important to remember that while the disease has an overall progression, the course will vary between patients. Some symptoms may worsen, while others may fluctuate in their severity.13
Determining a Huntington’s disease diagnosis
Huntington’s disease is currently diagnosed through a combination of clinical assessments and genetic testing.2,10,12
Patient history and clinical assessments
The preliminary diagnostic work-up of a suspected case of Huntington’s disease involves questionnaires, a general physical exam, a review of family medical history, and neurological and psychiatric examinations.7,14
Diagnostic imaging
Brain imaging (magnetic resonance imaging, positron emission tomography, or computed tomography) may also be performed to assess the structure or function of the brain. These images may reveal structural, functional, and molecular changes in areas of the brain affected by Huntington’s disease; however, such changes may not be seen if imaging is performed early in the course of the disease.7,14,15
Genetic tests
Genetic testing for the presence of the HTT mutation is conducted in most instances.16
In both predictive and diagnostic circumstances, genetic testing can be used to confirm Huntington’s disease in cases where typical symptoms are displayed.16
What is the prognosis of Huntington’s disease?
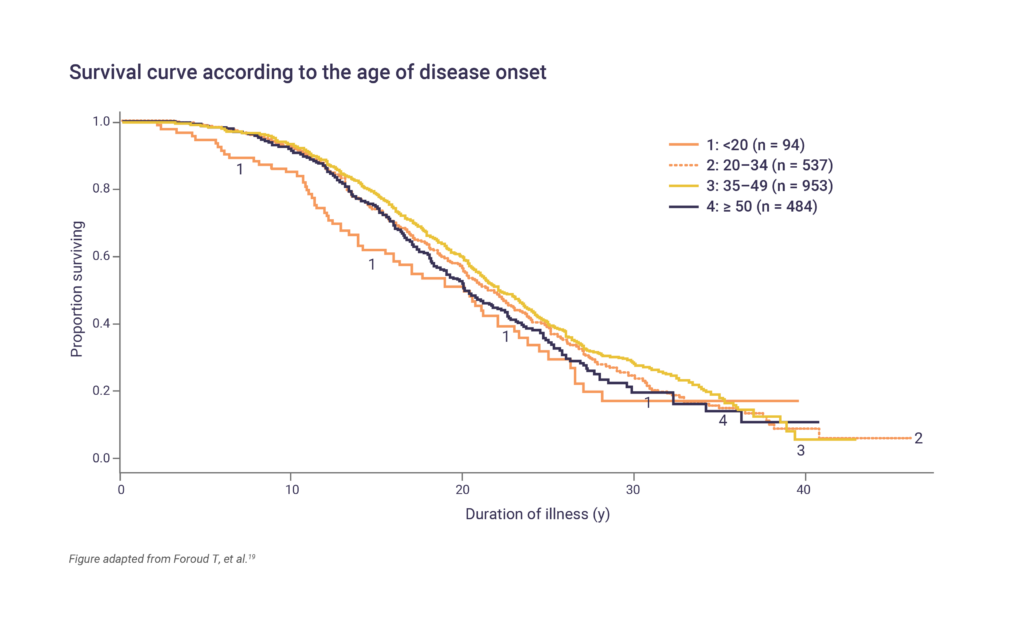
Huntington’s disease progresses slowly over 15–20 years following the onset of symptoms.17 The prognosis and life expectancy of patients with Huntington’s disease can vary depending on age of onset, the severity of symptoms and rate of disease progression, as well as other factors. Pneumonia and injuries related to falls are the most common causes of death.10,18 People who develop symptoms at a younger age, including those diagnosed with Juvenile Huntington’s disease, may have a more rapid progression of the disease and shorter life expectancy compared with those with an onset between 20 and 49 years.19
CAG, cytosine, adenine, and guanine.
References
- World Health Organization, 2020. 8A01.10 Huntington disease. Available at: https://icd.who.int/browse11/l-m/en#/http://id.who.int/icd/entity/2132180242. Accessed August 2023.
- Caron NS, Wright GEB, Hayden MR. Huntington’s Disease. 1998 Oct 23 [Updated 2020 Jun 11]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews®. US, 1993–2020.
- Ranganathan M, Kostyk SK, Allain DC, et al. Clin Genet 2021;99:133–145.
- Huntington Study Group. HD Basics. Available at: https://huntingtonstudygroup.org/living-with-hd/hd-basics/. Accessed August 2023.
- Huntington’s Disease Society of America. Overview of Huntington’s Disease. Available at: https://hdsa.org/what-is-hd/overview-of-huntingtons-disease/. Accessed August 2023.
- Gatto EM, González Rojas N, Persi G, et al. Clin Parkinsonism Rel Disord 2020;3:100056.
- Baig SS, Strong M, Quarrell OW. Neurodegener Dis Manag 2016;6:331–343.
- Medina A, Mahjoub Y, Shaver L, Pringsheim T. Mov Disord 2022;37(12):2327–2335.
- McColgan SJ, Tabrizi P. Eur J Neurol 2018;25:24–34.
- Roos RAC. Orphanet J Rare Dis 2010;5:40.
- Kirkwood SC, Su JL, Conneally P, et al. Arch Neurol 2001;58:273–278.
- Paulsen JS. Curr Neurol Neurosci Rep 2011;11:474–483.
- Rosenblatt A. Chapter 1: Overview and Principles of Treatment. eds. In: Nance M, et al. eds. A Physician’s Guide to the Management of Huntington’s Disease. US; 2011.
- Mayo Clinic. Huntington’s disease. Available at: https://www.mayoclinic.org/diseases-conditions/huntingtons-disease/diagnosis-treatment/drc-20356122 Accessed August 2023.
- Roussakis A and Piccini P. J Huntingtons Dis 2015;4(4):287–296.
- Bates GP, et al. Nat Rev Dis Primers 2015;1:1–21.
- Craufurd D, MacLeod R, Frontali M, et al. Pract Neurol 2015;15:80–84.
- National Institute of Neurological Disorders and Stroke, 2020. Huntington’s Disease: Hope Through Research. Available at: https://catalog.ninds.nih.gov/sites/default/files/publications/huntingtons-disease-hope-through-research.pdf. Accessed June 2024.
- Foroud T, Gray J, Ivashina J, et al. J Neurol Neurosurg Psychiatry 1999;66:52–56.